病態・定義
遺伝性痙性対麻痺は臨床上は緩徐進行性の下肢痙縮と筋力低下を認め、病理学的には脊髄の錐体路、後索、脊髄小脳路の変性を認める神経変性疾患です。多くは常染色体優性遺伝で、一部常染色体劣性遺伝などが存在し、SPG〇〇と遺伝子に番号がふられています。細胞内輸送の障害(小胞体など)が病態の主体とされていますが、なぜそれが痙性対麻痺の臨床像を呈するのかはまだ解明されていません。
臨床的には痙性対麻痺のみを呈する純粋型(pure form)(軽度の深部感覚障害と膀胱障害はpure formに含まれます)とこれに加えてその他の臨床症候(末梢神経障害、小脳失調、精神発達遅滞、痙攣、難聴、網膜変性症、魚鱗癬、症状ではないが画像所見など)を伴う複合型(complicated form)に分類されます。一般的に純粋型はADが多く、複合型はARやX連鎖性に多いとされています。(参照:「脊髄小脳変性症・多系統萎縮症診療ガイドライン2018」)。
日本でのAD遺伝形式ではJASPACからは206家系中SPG4(78家系:38%)が最も多く、SPG3A(11家系:5%)、SPG31(10家系:5%)、SPG10(3家系:2%)、SPG8(1家系:1%)と報告されている。
またAR遺伝形式の116例解析ではではSPG11(14例:12%)が最も多く、続いてSPG28(5例:4%)、SPG46(4例:3%)、SPG15(3例:3%)、SPG7,21,35,52,54,56,57が1例ずつと報告されています。
複合型(complicated form)の代表例
・白質病変:SPG5, 7, 21, 35
・下位運動ニューロン障害:SPG10, 11, 17, 20, 39
・脳梁菲薄化:SPG11
・末梢神経障害:SPG3A
臨床像
下肢の痙性による歩行障害を認めますが、歩行障害に比して筋力低下はあまり目立たない(特に病初期には)ことが特徴的と思います(多発性硬化症や脊髄炎では痙性とともに筋力低下も目立つことと比べて)。
痙性に特異的な病歴を拾うという作業は非常に難しく、病歴だけから「痙性」を判定することは難しいです。神経診察でspasticityをきちんと拾う作業が出来ればアプローチは比較的容易と思います。
変性疾患らしく、非常に緩徐な経過をたどることも特徴です。
痙性により足関節が底屈した状態が基本となるため、つま先が地面にすってしまい靴のつま先側がすり減るという特徴があります。私は毎回靴を観察するようにしています(以下は自験例痙性対麻痺患者さんの靴の写真)

臨床評価スケール:Spastic Paraplegia Rating Scale (SPRS) 0~42点で評価(点数が大きいほど重症)
痙性の評価:modified Ashworth Scale (MAS) 0~4点で評価
■ドイツ家系遺伝性痙性対麻痺608例のまとめ ANN NEUROL 2016;79:646–658 *重要論文
HSP608例の内訳は下記の通り:SPG4 196例、SPG7 28例、SPG11 15例、SPG5 10例、SPG3 9例、SPG15 7例、SPG10 5例、SPG31 5例、SPG17 3例、SPG21 2例、SPG35 2例、SPG39 2例、SPG1 1例、SPG2 1例、SPG8 1例、SPG28 1例、SPG46 1例、SPG58 1例、その他9例、不明309例。
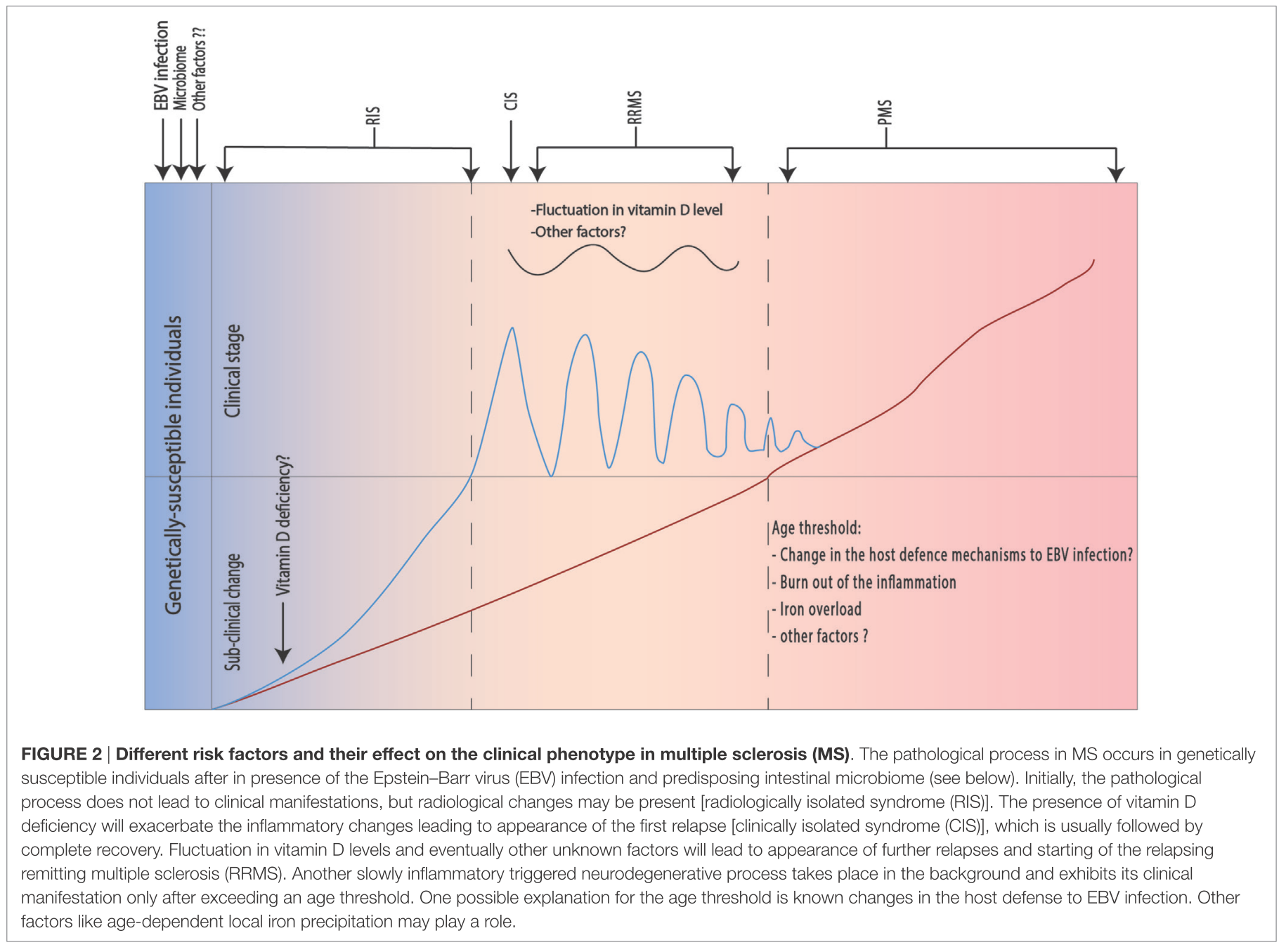
発症年齢は平均30.8歳(0-73歳)、分布は2峰性で5歳未満と40歳前後にピークがあります(下図参照)。
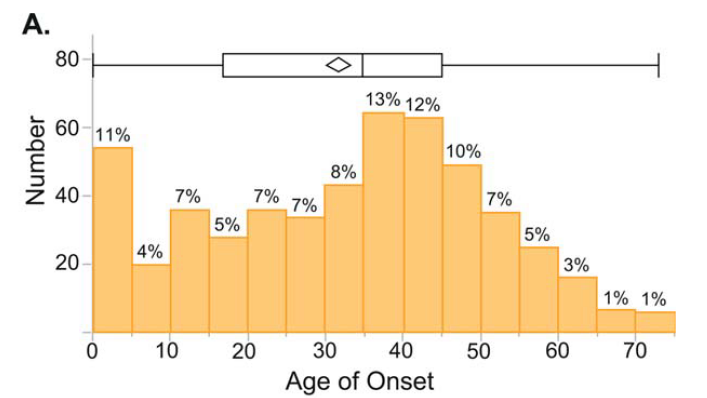
SPGごとの発症年齢の違いは下図の通りです。
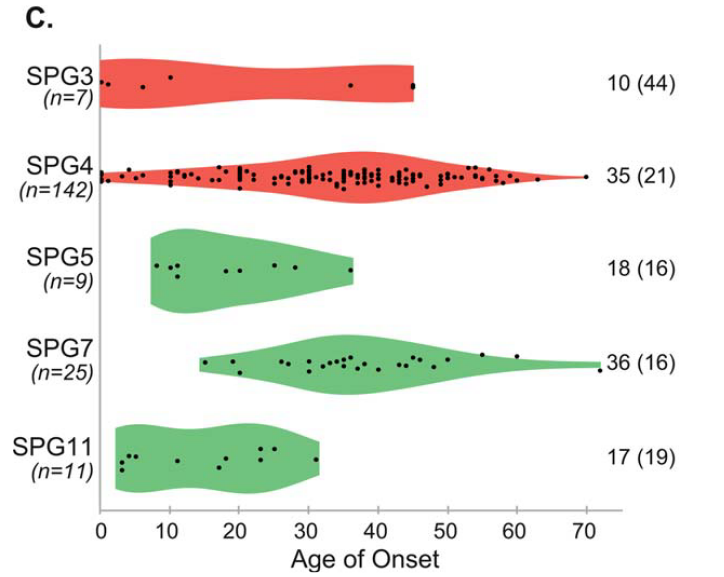
錐体路徴候以外の臨床症候は75%に認め、感覚障害>50%、失調28%、下位運動ニューロン障害19%に認め、図にまとめると下記の通りです。
予後に関しては発症後22年で半数が歩行補助具が必要となり、37年で約1/4で車いすが必要とされています。高齢発症では経過は重症であり早期に介助歩行となることが指摘されています。発症年齢が遅い症例は進行が早いことと、認知機能障害、嚥下障害、錐体外路症状、下位運動ニューロン障害などが重症度と関連しています。
鑑別診断
特に20歳以降の発症で家族歴がなく痙性対麻痺を呈する患者の場合、遺伝性痙性対麻痺はあくまで除外診断なので注意が必要です。
■小児発症の場合:脳性麻痺、Leukodystrophy(Krabbe’s diseaseなど)、環軸堆亜脱臼、Chiari奇形、無βリポプロテイン血症、多発性硬化症、Levodopa-responsive dystonia
■成人発症の場合:多発性硬化症、MND、頚椎症性脊髄症、腫瘍、髄膜腫(傍正中)、脊髄炎、DAVF、Chiari奇形、副腎白質ジストロフィー、ビタミン欠乏症(B12, E)、銅欠乏、感染症(梅毒、HTLV1、HIV)、銅欠乏症、Levodopa-responsive dystonia
*頚椎症性脊髄症に関してはこちらをご参照ください。
*PPMSに関してはこちらをご参照ください。
*HTLV-1関連脊髄症(HAM/TSP)に関してはこちらをご参照ください。
検査
以下の検査項目を痙性対麻痺を呈する疾患の鑑別で検討します。
血液検査:ビタミンB12, E、リポプロテイン分画、コレスタノール、銅、セルロプラスミン、超長鎖脂肪酸
感染症関連:梅毒、HTLV1、HIV
髄液検査:一般的な項目OCB(特に脊髄炎やPPMS)、細胞診
画像:脊髄MRI検査(特に脊髄萎縮・信号変化、dAVFらしいflow voidにも注意)・頭部MRI検査(特に脳梁菲薄化・錐体路信号変化・Chiari奇形、その他鑑別として多発性硬化症の所見がないかどうか?)
電気生理検査:神経伝導検査、針筋電図(下位運動ニューロン障害合併がないかどうか)、SEP、MEP(可能なら実施を検討)
眼科:特にMS(PPMS)を鑑別として疑う場合(OCT)
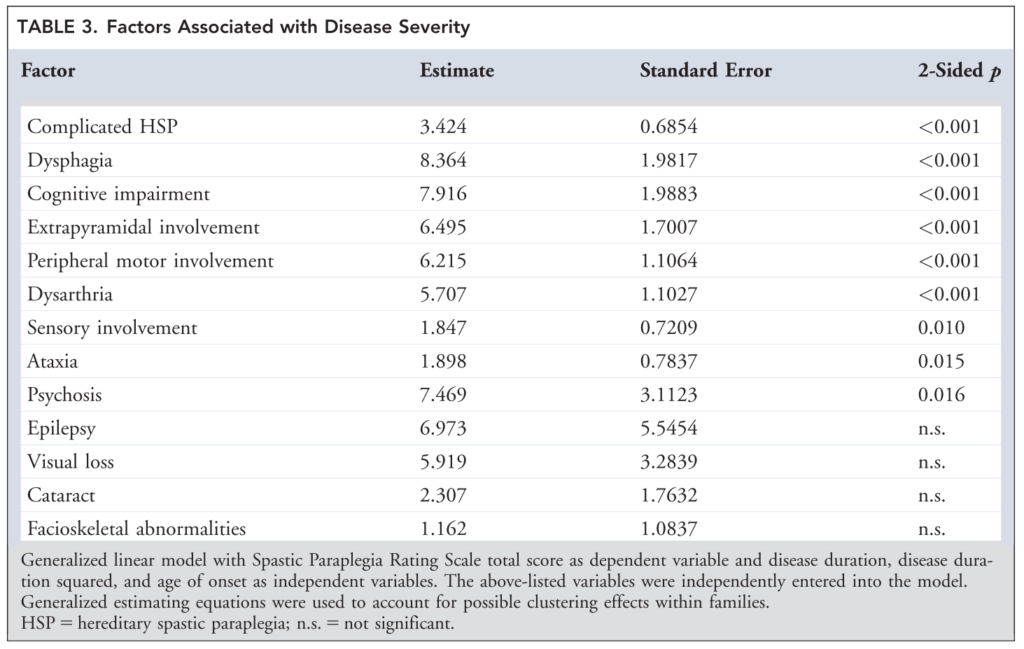
■AD-HSP(SPG3A, SPG4, SPG6,SPG8)の13例の脊髄MRI所見の特徴 Neuroradiology (2005) 47: 730–734
同年代の健常者と比較して全例で脊髄萎縮をAD-HSPでは認めています(下図参照)。脊髄萎縮は頚髄よりも胸髄領域で特に有意に認めています。AD-HSPの中では特にSPG6, 8で脊髄萎縮をSPG3,4よりも高度に認めたと報告されていますSPG6, SPG8 (47.60±6.58 mm2 at C2, 21.40±2.4 mm2 at T9) はSPG3,SPG4(66.0±8.94 mm2 at C2, p<0.02;31.75±2.76 mm2 at T9, p<0.001)。
各論
SPG11
・常染色体劣性遺伝形式(ARの中で最も頻度が高い)、15染色体長腕(15q21.1)に存在するSPG11遺伝子変異(Nat Genet 2007;39:366)
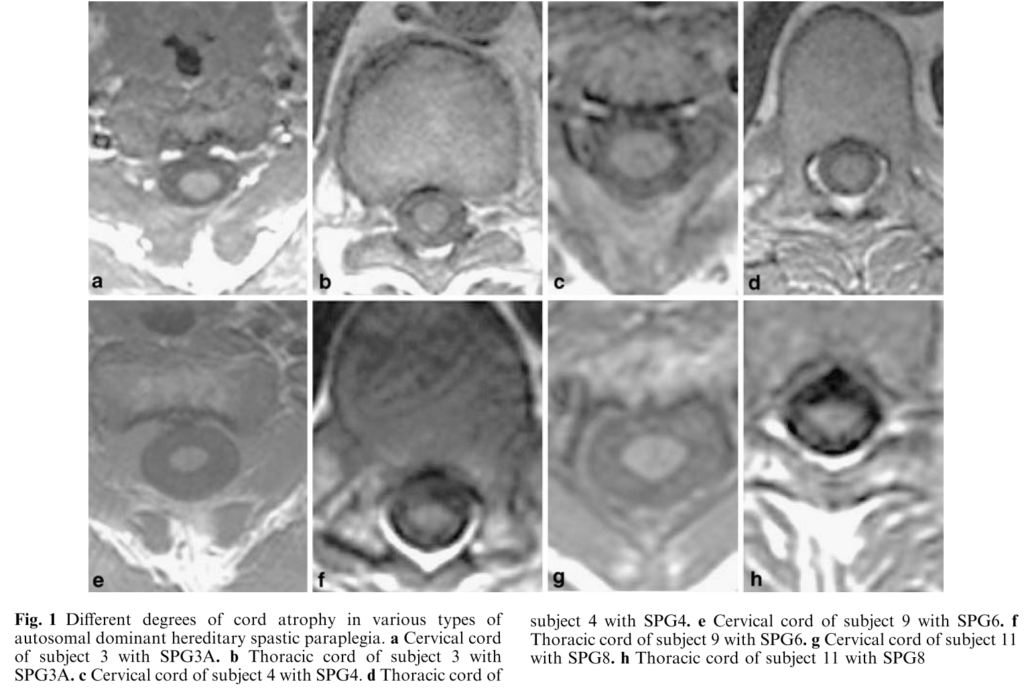
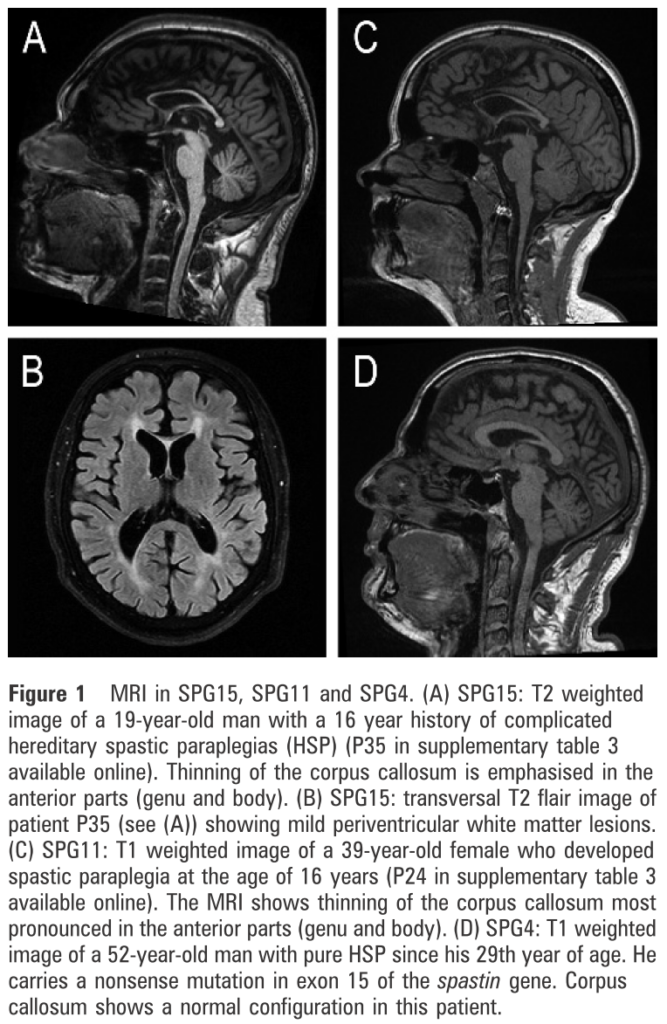
・精神発達遅滞、画像所見で脳梁菲薄化が特徴的
■SPG11の38例まとめ Brain (2008), 131, 772^784
臨床像:発症年齢は2-77歳(14.0±5.9歳)、初発症状は歩行障害79%、精神発達遅滞16%を認めています。その他の臨床症状としては小脳性眼球運動障害(saccade運動障害や眼振)、pes cavus、側彎症、その他パーキンソニズム、起立性低血圧、黄斑変性、斜視などを伴う場合もあります。
電気生理検査:81%(13/16)で下位運動ニューロン障害の所見を認めたとされています。萎縮(53%)が強くなるとMND mimicとなる場合もあり注意が必要です。
画像所見:脳梁菲薄化(thin corpus callosum:TCC)95%(20/21)、皮質萎縮81%(17/21)、白質病変69%(13/19)(白質病変は軽度の場合は前角・後角周囲白質のみであるが経過とともに増悪する)が特徴的とされています(下図参照)。
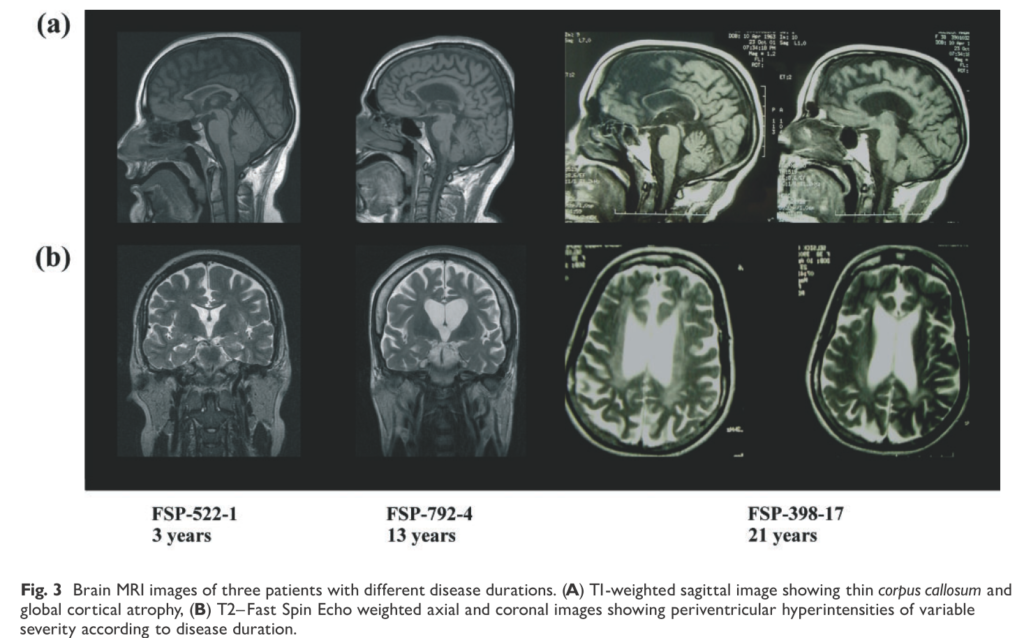
経過と機能予後は以下の通りで発症してから平均16.5年で車椅子移動のADLとなります。
また本疾患は脳梁菲薄化(TCC)を伴った痙性対麻痺の中では最も多いことが分かります。
■SPG15はSPG11の次に脳梁菲薄化を伴うHSPとして頻度が多い Neurology ® 2009;73:1111–1119
SPGはAR-HSPの中(ヨーロッパ、地中海周辺の地域に多い)で精神発達遅滞、黄斑変性症、遠位の筋萎縮とされていましたが、ZFYVE26の変異が報告され、脳梁菲薄化を伴う症例もあることが分かりました。ここでは8家系11人のSPG15患者に関してまとめています。
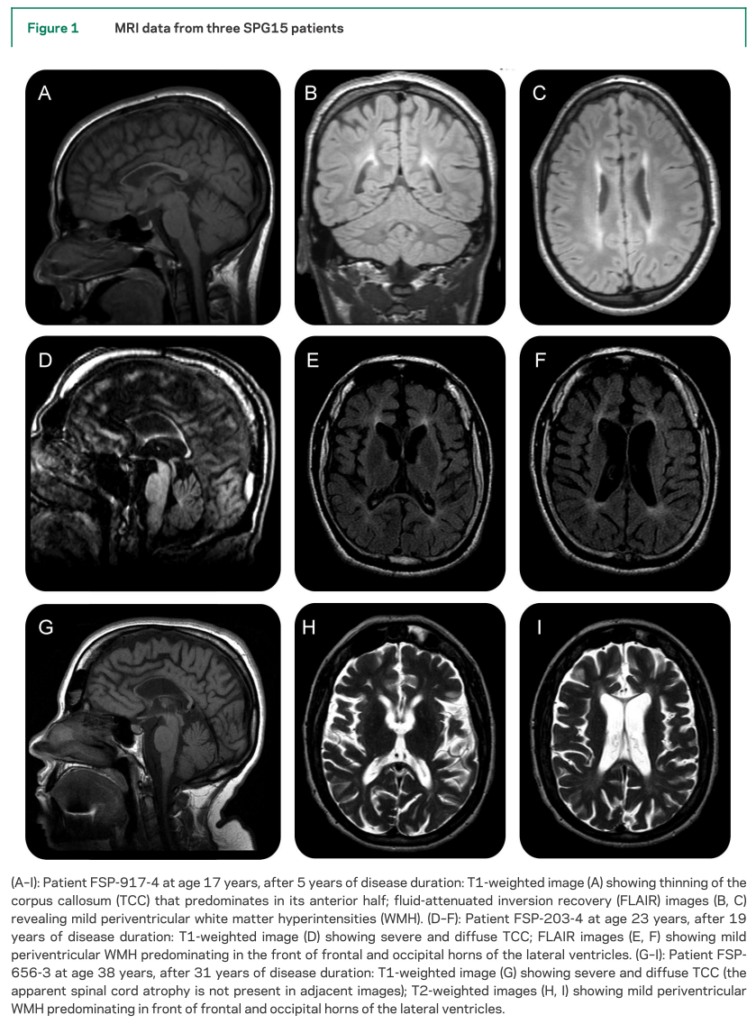
SPG11とSPG15の既報例に関して違いを検討したものは下図になります。
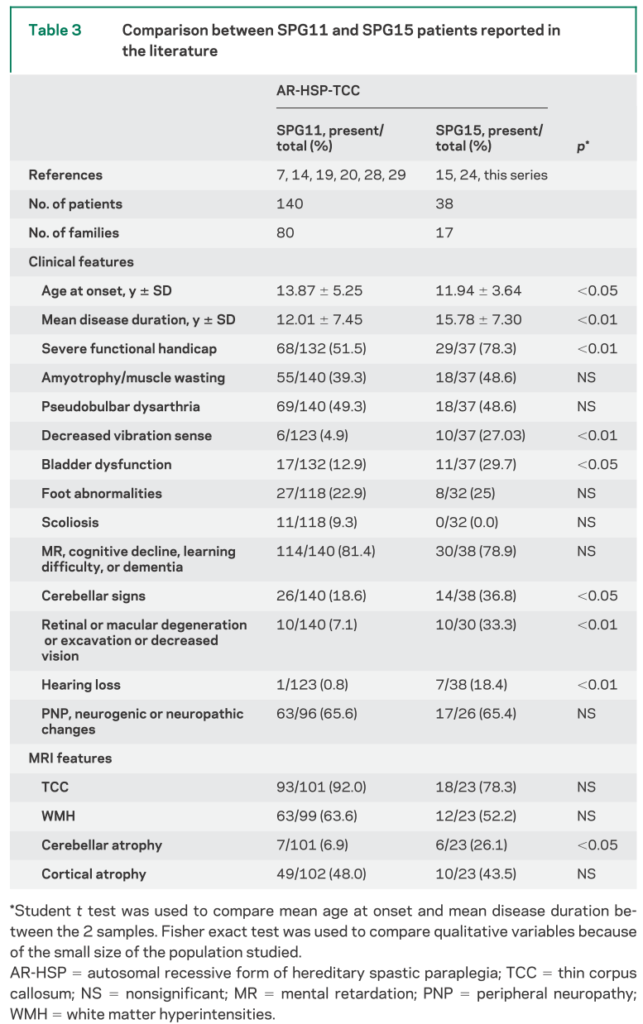
■SPG11とSPG15は臨床的に区別することは困難 J Neurol Neurosurg Psychiatry 2009;80:1402–1404.
36例早期発症のAR-HSP(complicated form)でSPG11, 15を検索し、SPG11は14%、SPG15は2.6%に認めた。脳梁菲薄化を伴うHSPはSPG11, SPG15を示唆するが、SPG11とSPG15両者を区別する臨床兆候は指摘することが出来なかった。
治療
根本的な治療法はなく痙縮に対する対症療法が基本となります。
筋弛緩剤
・中枢性筋弛緩剤:チザニジン、バクロフェン、クロナゼパムなど
・末梢性筋弛緩剤:ダントロレン
その他:ボツリヌス毒素筋注、モーターポイントブロック、バクロフェン持続髄注療法(ITB)など
参考文献
・Lancet Neurol 2008; 7: 1127–38